Strefa okazji
Strefa okazji
 Nowości
Nowości
 Krótkie daty
Krótkie daty





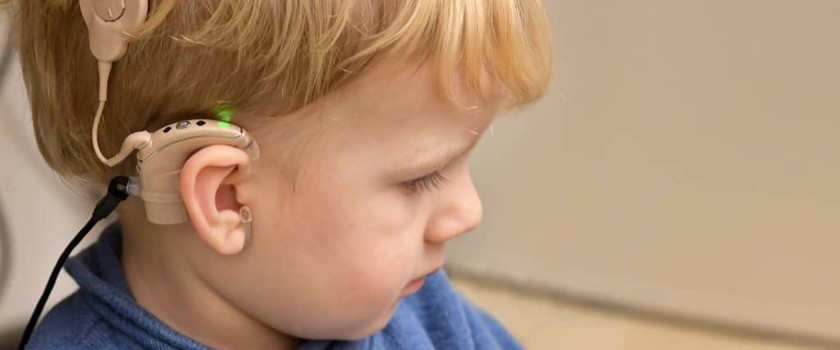

Czym jest zespół Ushera? Badanie niemieckich naukowców daje nowe spojrzenie na patologię choroby
Zespół Ushera to choroba dziedziczona genetycznie od obojga rodziców. Oznacza to, że zarówno matka, jak i ojciec przekazują dziecku po jednej kopii wadliwego genu, co tylko w takiej konfiguracji będzie skutkowało wystąpieniem tzw. głuchoto-ślepoty. Obecny stan wiedzy nie pozwala na doraźne leczenie tej choroby z użyciem środków farmakologicznych, a jedynie kompensacje wady słuchu z użyciem implantu ślimakowego. Najnowsze wyniki badań naukowych pozwalają jednak dostrzec światełko w tunelu, ponieważ jak donoszą niemieccy naukowcy, właśnie odkryto patologiczny mechanizm molekularny, prowadzący do rozwoju choroby, a to w przyszłości może pozwolić na opracowanie skutecznej metody leczenia zespołu Ushera, z wykorzystaniem np. terapii genowej.
Naukowcy z Instytutu Fizjologii Molekularnej uniwersytetu Johannesa Gutenberga w Moguncji zidentyfikowali właśnie nowy mechanizm genetyczny prowadzący do wywołania objawów zespołu Ushera. To pierwsze dowody na to, że dysregulacja procesu splicingu może uczestniczyć w patofizjologii tej choroby.
Czym jest zespół Ushera?
Zespół Ushera (UCH, ang. Human Usher syndrome) jest najczęstszą postacią dziedzicznej głucho-ślepoty. Dotyka jedną na 6000 osób na całym świecie. Większość dzieci z zespołem Ushera rodzi się z deficytem słuchu w stopniu umiarkowanym, a nawet głębokim. Typ głuchoty często jest także określany jako odbiorczy, a zwyrodnienie oka dotyczy barwników siatkówki (łac. retinitis pigmentosa). Oprócz tego mogą występować jeszcze inne poważne zaburzenia, takie jak brak funkcji przedsionka serca, opóźnienie rozwoju psychomotorycznego lub ciężki niedorozwój mowy. Ubytek rzadziej pojawia się w okresie dojrzewania i później. Pacjenci mogą także cierpieć z powodu zaburzeń równowagi oraz tracić wzrok w miarę postępu choroby. Farmakologiczne leczenie zespołu Ushera jest obecnie niemożliwe, ponieważ nie istnieje żaden preparat skutecznie zwalczający głuchoto-ślepotę. Implanty ślimakowe mogą być stosowane jedynie w celu skompensowania ubytku słuchu.
Zespół Ushera – genetyczne podstawy choroby
Każda osoba dziedziczy dwie kopie genu, po jednej od każdego rodzica. Zmutowane geny mogą powodować nieprawidłowy rozwój lub działanie komórek. Kiedy dziecko dziedziczy dwie kopie wadliwego genu, rozwijają się objawy zespołu Ushera. Osoba z jednym nieprawidłowym genem odpowiedzialnym za wywołanie syndromu nie ma zaburzenia, ale jest nosicielem. Warto dodać, że choroba powstaje w wyniku mutacji jednego z 10. genów typowych dla tej choroby.
Zespół Ushera jest dziedziczony:
- autosomalnie – zmutowany gen nie znajduje się na żadnym z chromosomów, które określają płeć danej osoby, dlatego zarówno mężczyźni, jak i kobiety mogą mieć zaburzenia i przekazać je potomstwu;
- recesywnie – osoba musi otrzymać zmutowaną formę genu od każdego rodzica (nie wystarczy pojedyncza kopia genu), aby rozwinąć objawy choroby.
Powiązane produkty
-

wyrób medyczny, spray, korek woskowinowy
24,19 złDo koszyka -
wyrób medyczny, ampułka, korek woskowinowy
14,59 złDo koszyka -
wyrób medyczny, krople, korek woskowinowy, zapalenie
22,59 złDo koszyka -
Bestseller
wyrób medyczny, spray, korek woskowinowy
19,99 złDo koszyka -
wyrób medyczny, spray, korek woskowinowy
22,79 złDo koszyka

Patologia zespołu Ushera — nowe ustalenia
W ciągu ostatnich 25. lat grupa kierowana przez Uwe Wolfruma profesora w Instytucie Fizjologii Molekularnej Uniwersytetu Johannesa Gutenberga w Moguncji (Niemcy) badała mechanizmy leżące u podstaw zespołu Ushera. Wraz ze współpracownikami z Instytutu Chemii Biofizowej Maxa Plancka w Getyndze, udało się zidentyfikować patologiczny mechanizm prowadzący do rozwoju choroby.
Naukowcy odkryli, że białko SANS, którego synteza warunkowana jest działaniem genu USH1G, odgrywa kluczową rolę w regulacji molekularnego procesu zachodzącego w komórce, zwanego splicingiem (złożony mechanizm składania genów). Co więcej, wskazali, że defekty w białku SANS mogą prowadzić do błędów w splicingu genów związanych z zespołem Ushera, co może wywołać chorobę.
Wcześniejsze badania Wolfruma wykazały, że SANS działa jak rusztowanie. Ma wiele domen, do których mogą dokować (przytwierdzać się, przyłączać) inne białka, dzięki czemu umożliwia prawidłowe funkcjonowanie komórki. Tym samym mutacje białka SANS mogą prowadzić do wystąpienia szeregu kaskadowych nieprawidłowości molekularnych, skutkujących zaburzeniami w funkcjonowaniu komórek słuchowych, przedsionkowych komórek narządu Cortiego w uchu wewnętrznym i fotoreceptorowych komórek siatkówki oka, powodujących trudności odbioru bodźców sensorycznych.
Profesor Wolfrum stwierdził, że oprócz nowych ustaleń związanych z mechanizmem splicingu, udało się zidentyfikować także nowe aspekty, które wymagają jeszcze zbadania, ale prawdopodobnie w przyszłości, okażą się one znaczące dla opracowania koncepcji leczenia zespołu Ushera.
Czym jest splicing genu? Jakie jest jego znaczenie dla rozwoju zespołu Ushera?
Splicing jest istotnym procesem na drodze biosyntezy białek. W biologii termin splicing oznacza składanie genu, które polega na usunięcie intronów, (sekwencji niekodujących) ze wstępnie transkrybowanego pre-mRNA lub w przypadku splicingu alternatywnego wykluczenie eksonów (odcinków kodujących sekwencje aminokwasów w cząsteczce białka), które nie są wymagane dla kolejnego wariantu. Proces splicingu jest katalizowany w jądrze przez spliceosomy, tj. dynamiczną, wysoce złożoną maszynę molekularną, która jest sukcesywnie składana ze składników białkowych i RNA.
Brak białka SANS, a także patogenne mutacje genu USH1G/SANS uniemożliwiają prawidłowe składanie i sekwencyjną aktywację spliceosomu. To z kolei hamuje prawidłowe składanie innych genów związanych z zespołem Ushera, prowadząc ostatecznie do ich dysfunkcji, a tym samym do rozwoju zaburzenia.