 Strefa okazji
Strefa okazji
 Nowości
Nowości
 Krótkie daty
Krótkie daty






Czym są priony? Jakie choroby wywołują i jak się je leczy?
Choroby prionowe dotykają zarówno ludzi, jak i zwierzęta. Są wywoływane przez nieprawidłowo sfałdowane białka transmisyjne – akumulacja odbiegających od normy protein może powodować wiele dotkliwych objawów, które z czasem prowadzą do śmierci. Jakie są rodzaje chorób prionowych? Czy jest sposób, aby im zapobiec?
Czym są priony? Jaka jest różnica między prionem prawidłowym i patologicznym?
Priony są specyficznymi cząsteczkami białkowymi, do których przyłączona jest reszta cukrowa i kwas sialowy. Są prostymi polipeptydami zbudowanymi z około 250 aminokwasów, o masie cząsteczkowej od 30 do 37 kDa, w warunkach fizjologicznych znajdują się na powierzchni komórek. Na skutek zmiany konformacji białka, prawidłowe priony (ang. cellular prion protein, PrPc) mogą przybierać inną strukturę konformacyjną i przekształcać się w patologiczne izoformy (ang. scrapie isoform of prion protein, PrPsc), które stanowią czynnik chorobotwórczy i uczestniczą w tworzeniu się amyloidu. W komórce można je znaleźć w endosomach i lizosomach.
Przyczyny chorób prionowych
Choroby prionowe dzieli się na dziedziczne oraz nabyte. Wyróżnia się także tzw. sporadyczne zakażenia.
- Dziedziczne choroby prionowe – są wywołane są mutacjami w genie PRNP, który zawiera instrukcje dotyczące wytwarzania białek prionowych.
- Nabyte choroby prionowe – są wynikiem ekspozycji na PrPSc ze źródła zewnętrznego, gdy normalne białko (PrPc) spontanicznie, z nieznanych przyczyn, przekształca się w nieprawidłowo ukształtowanego białka (PrPSc). Patologiczne białka gromadzą się w mózgu, tworząc grudki, które uszkadzają neurony i promują powstawanie wakuol, co prowadzi do powstawania charakterystycznych gąbczastych zmian mózgowia i wywołania objawów choroby prionowej.
- Sporadyczne zakażenia – są wywołane przez przypadkowe narażenie na tkankę skażoną PrPSc podczas zabiegu medycznego, np. przeszczepu rogówki. Zdarzają się one incydentalnie.
Powiązane produkty
-

kapsułki, odporność, niedobór witamin, witamina D
17,99 złDo koszyka -
syrop, malinowy
14,99 złDo koszyka -
tabletka, odporność, zmęczenie, alergia, niedobór minerałów, osłabienie, złe samopoczucie
5,99 złDo koszyka -
syrop, czarny bez, kwiaty
14,99 złDo koszyka -
sok NFC, certyfikowany ekologicznie
34,99 złDo koszyka -
7,99 złDo koszyka
-
saszetki, owocowy, żurawina
6,99 złDo koszyka







Choroby prionowe u ludzi
Dokładna częstość występowania chorób prionowych nie jest znana, ale są one rzadkie. Szacuje się, że na całym świecie każdego roku spotykają jedną na milion osób. Wspólnie nazywa się je pasażowalnymi encefalopatiami gąbczastymi (TSE, ang. transmissible spongiform encephalopathies). Cechują się one wieloletnim okresem inkubacji i w początkowej fazie trudno je rozpoznać. Rozwojowi choroby i destrukcyjnym zmianom w mózgu towarzyszą objawy neurologiczne, psychiatryczne oraz otępienie, a także zaburzenia ruchu. Obecnie choroby prionowe są nieuleczalne, ale wciąż poszukiwane są sposoby leczenia tych schorzeń.
Jednostki chorobowe diagnozowane u ludzi to:
- Choroba Creutzfelda-Jakoba (CJD) – choroba neurodegeneracyjna mózgu, która rozwija się w wyniku zadziałania czynnika zewnętrznego. Powoduje gąbczaste zwyrodnienie mózgu i stopniowo prowadzi do utraty umiejętności wykonywania codziennych czynności. Okres inkubacji wynosi zazwyczaj od kilku do kilkunastu lat, a śmierć pacjenta następuje do kilkunastu miesięcy od wystąpienia pierwszych objawów klinicznych. Choroba jest niejednorodną jednostką i przysparza trudności diagnostycznych. Ze względu na etiologię wyróżnia się cztery postacie choroby: samoistną (sCJD), rodzinną (fCJD), jatrogenną (jCJD) oraz wariant związany z przeniesieniem choroby wściekłych krów na człowieka (vCJD).
- Choroba Gerstmanna-Sträusslera-Scheinkera (GSS) – to niezwykle rzadka, genetycznie uwarunkowana choroba zakaźna, objawiająca się postępującą ataksją móżdżkowo-rdzeniową z towarzyszącymi jej objawami otępienia.
- Choroba Kuru– została zidentyfikowana w populacji South Fore w Papui Nowej Gwinei. Do zakażenia dochodziło, gdy ludzie zjadali dotkniętą ludzką tkankę podczas kanibalistycznych rytuałów pogrzebowych. Spowodowała śmierć blisko 90% ludności tej społeczności. Ze względu na większą świadomość choroby, obecnie jest ona rzadkością.
- Śmiertelna rodzinna bezsenność (FFI) – bardzo rzadka autosomalna choroba dziedziczna, powodująca trudności w zasypianiu. Po dłuższym czasie chorobliwy brak snu może doprowadzić do wywołania ataków paniki, halucynacji i demencji. Śmierć następuje w ciągu kilku miesięcy do kilku lat od wystąpienia pierwszych objawów.
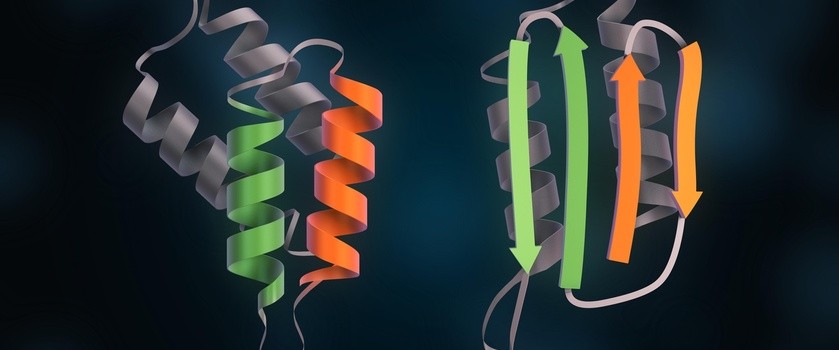
Odkryto mechanizm molekularny nieprawidłowego fałdowania prionów
Molekularne mechanizmy leżące u podstaw nieprawidłowego fałdowania białek prionowych i sprzyjające ich agregacji nie są dobrze poznane. Naukowcom z Imperial College London i Uniwersytetu w Zurychu udało się jednak niedawno ustalić, co powoduje, że normalne białka prionowe przekształcają się w formę chorobotwórczą. Wykorzystując spektroskopię rezonansu jądrowego w połączeniu z analizą obliczeniową, zlokalizowali strukturę etapu pośredniego podczas procesu zmierzającego do powstania infekcyjnego białka i znaleźli sposób na jego zablokowanie, co może prowadzić do powstania nowych terapii.













